https://blog.tensorflow.org/2020/08/optimizing-peptides-in-tensorflow-2.html?hl=he
https://blogger.googleusercontent.com/img/b/R29vZ2xl/AVvXsEiXHunNqbwAf6OPZlFQTEXiWKL_-bdVQJRsKZWeeYWfmiAMQFmx3zST_9U5n8OYNdOpQBAMHWjTijIDh0xnvzrdnNyhe6R5Wkz8J5NxaCPmb-Ipu27lmi0xTyDtCI3SnNRhBXssHmpIt50/s1600/CPP.jpg
A guest post by Somesh Mohapatra, Rafael Gómez-Bombarelli of MIT
Introduction
A polymer is a material made up of long repeating chains of molecules, like plastic or rubber. Polymers are made up of subunits (monomers) that are chemically bound to one another. The chemical composition and arrangement of monomers dictate the properties of the polymer. A few examples of polymers in everyday use are water bottles, non-stick teflon coatings, and adhesives.
 |
| Figure 1. Conceptually, you can think of Peptimizer as generating a sequence of amino acids, then predicting a property of the peptide, then optimizing the sequence. |
Peptides are short polymer chains made up of amino acids, analogous to words composed of letters. They are widely used for therapeutic applications, such as for the delivery of gene therapy by cell-penetrating peptides. Thanks to their modular chemistry amenable to automated synthesis and expansive design space, peptides are increasingly preferred over more conventional small molecule drugs, which are harder to synthesize. However, the vast sequence space (in terms of the amino acid arrangement) acts as an impediment to the design of functional peptides.
Synthetic accessibility, apart from functionality optimization, is a challenge. Peptides and other functional polymers with a precise arrangement of monomers are synthesized using methods such as flow chemistry. The synthesis involves monomer-by-monomer addition to a growing polymer chain. This process necessitates a high reaction yield for every step, thus making the accessibility of longer chains challenging.
Conventional approaches for optimization of functional polymers, such as peptides, in a lab environment involve the heuristic exploration of chemical space by trial-and-error. However, the number of possible polymers rises exponentially as mn, where m is the number of possible monomers, and n is the polymer length.
As an alternative to doing an experiment in a lab, you can design functional polymers using machine learning. In our work on optimizing
cell-penetrating activity and
synthetic accessibility, we design peptides using
Peptimizer, a machine learning framework based on TensorFlow. Conceptually, you can think of Peptimizer as generating a sequence of amino acids, then predicting a property of the peptide, then optimizing the sequence.
Peptimizer can be used for the optimization of functionality (other than cell-penetrating activity as well) and synthetic accessibility of polymers. We use topological representations of monomers (amino acids) and matrix representations of polymer chains (peptide sequences) to develop interpretable (attribute the gain in property to a specific monomer and/or chemical substructure) machine learning models. The choice of representation and model architecture enables inference of biochemical design principles, such as monomer composition, sequence length or net charge of polymer, by using gradient-based attribution methods.
Key challenges for applying machine learning to advance functional peptide design include limited dataset size (usually less than 100 data points), choosing effective representations, and the ability to explain and interpret models.
Here, we use a dataset of peptides received from our experimental collaborators to demonstrate the utility of the codebase.
Optimization of functionality
Based on our
work on designing novel and highly efficient cell-penetrating peptides, we present a framework for the discovery of functional polymers (Figure 1). The framework consists of a recurrent neural network generator, convolutional neural network predictor, and genetic algorithm optimizer.
The generator is trained on a dataset of peptide sequences using Teacher Forcing, and enables sampling of novel sequences similar to the ones in the training dataset. The predictor is trained over matrix representations of sequences and experimentally determined biological activity. The optimizer is seeded with sequences sampled utilizing the generator. It optimizes by evaluating an objective function that involves the predicted activity and other parameters such as length and arginine content. The outcome is a list of optimized sequences with high predicted activity, which may be validated in wet-lab experiments.
Each of these components can be accessed from the
tutorial notebook to train on a custom dataset. The scripts for the individual components have been designed in a modular fashion and can be modified with relative ease.
Optimization of synthetic accessibility
Apart from functionality optimization,
Peptimizer allows for the optimization of
synthetic accessibility of a wild-type sequence (Figure 2). The framework consists of a multi-modal convolutional neural network predictor and a brute force optimizer. The predictor is trained over experimental synthesis parameters such as pre-synthesized chain, incoming monomer, temperature, flow rate, and catalysts. The optimizer evaluates single-point mutants of the wild-type sequence for higher theoretical yield.
The choice of a brute force optimizer for optimization of synthetic accessibility is based on the linearly growing sequence space (
m x
n) for the variations of the wild-type sequence. This sequence space is relatively small in comparison to the exponentially growing sequence space (
mn) encountered in optimization of functionality.
This framework may be adapted for other stepwise chemical reaction platforms with in-line monitoring by specifying the different input and output variables and respective data types. It can be accessed using a
tutorial notebook.
 |
Figure 2. Outline of synthetic accessibility optimization.
|
Interpretability of models
A key feature of
Peptimizer is the gradient-based attribution for the interpretation of model predictions (Figure 3). Taking the gradient of the predicted activity with the input sequence representation, we visualize both positive and negative activations for each input feature. Fingerprint indices corresponding to substructures that positively contribute to the activity have higher activation in the heatmap. This activation heatmap is averaged along the topological fingerprints axis to find key substructures or chemical motifs that contribute positively/negatively to the predicted activity. Averaging over the monomer position axis, we obtain the relative contribution of each monomer to the predicted functionality of the polymer. These visualizations provide in-depth insight into sequence-activity relationships and add to the contemporary understanding of biochemical design principles.
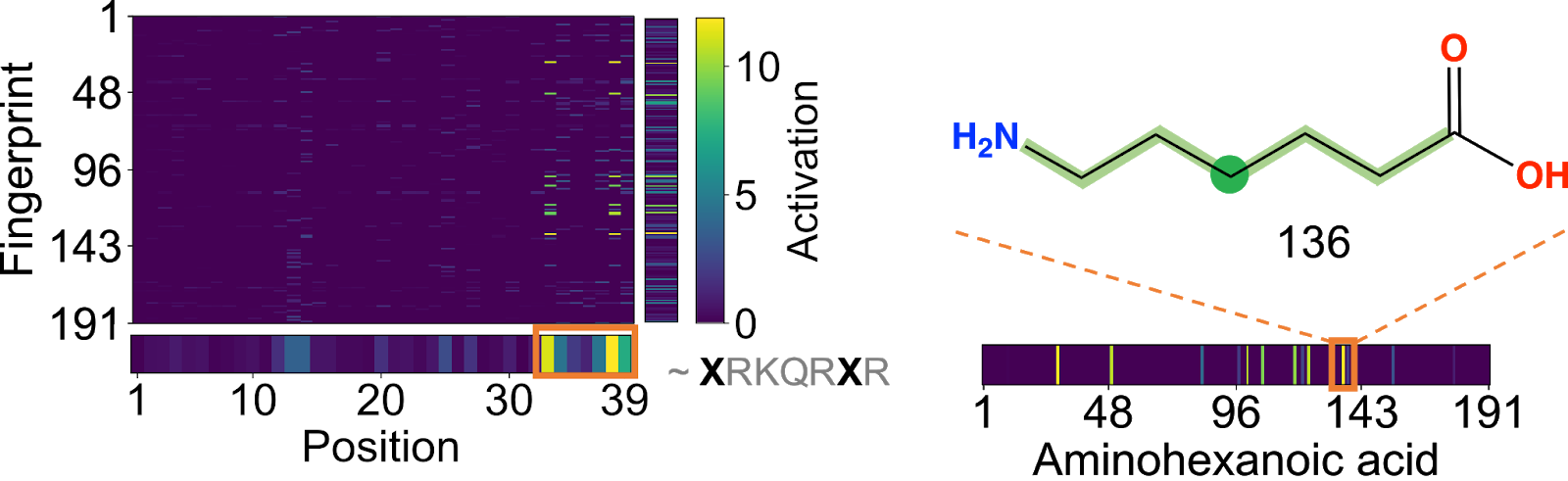 |
| Figure 3. (left) Positive gradient activation heatmap, and (right) activated chemical substructure, for functional peptide sequence. |
Outlook
Optimization of functional polymers using
Peptimizer can inform experimental strategies and lead to significant savings in terms of time and costs. We believe that the tutorial notebooks will help bench scientists in chemistry, materials science, and the broader field of sequence design to run machine learning models over custom datasets, such as
Khazana. In addition, the attribution methods will provide insights into the high-dimensional sequence-activity relationships and elucidation of design principles.
Experimental collaboration
This work was done in collaboration with the lab of Bradley Pentelute (Department of Chemistry, MIT). The collaborators for the optimization of functionality and synthetic accessibility were Carly Schissel and Dr. Nina Hartrampf, respectively. We thank them for providing the dataset, experimental validation, and the discussion during the development of the models.
Acknowledgment
We would like to acknowledge the support of Thiru Palanisamy and Josh Gordon at Google for their help with the blog post collaboration and with providing active feedback.





